Research
Single-cell CRISPR screens have emerged as a powerful tool for linking genetic perturbations (e.g. gene inhibition/activation by CRISPRi/a) to high-content measures of their phenotypic effects (e.g. the transcriptome or morphology). The Norman Lab played a foundational role in the development of one of these techniques, Perturb-seq, continues to develop new approaches that leverage it. Crucially, its transcriptome readout is both quantitative and interpretable, meaning it is possible to reason about progress towards realizing a desired phenotype using computational modeling. As it scales, we believe these features make Perturb-seq a promising tool for rationally engineering cellular transcriptional state.
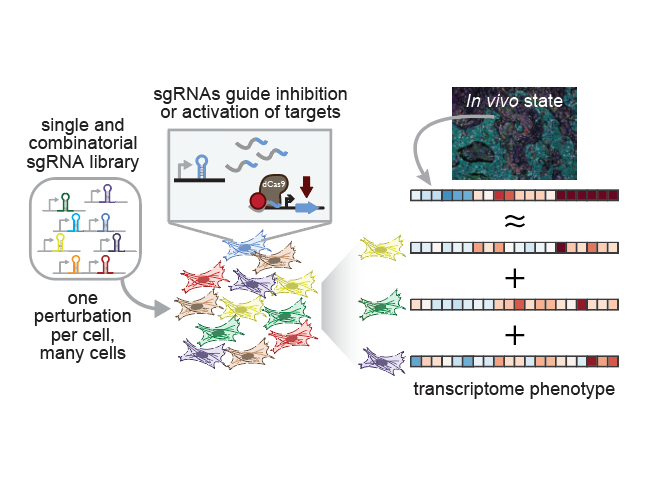
Engineering transcriptional state in vitro
Recent cell atlas studies have found remarkable diversity in cell types, but also revealed that even greater heterogeneity exists at the level of transcriptional states within tissues and across disease contexts. By trying to reconstruct these states in vitro we hope to understand their origins and learn principles for rationally engineering the transcriptomes of differentiated cell types, with eventual applications to engineering cell therapies.

Genetic drivers of fibroblast cell state
We are particularly interested in the biology of fibroblasts, which are ubiquitous throughout the body as the primary constituent cell type of connective tissue. They play critical roles in wound healing, fibrotic disease, and the tumor microenvironment. In vivo studies have identified fibroblast subsets defined by transcriptional markers that appear to have functionally distinct roles, some of which we have been able to elicit in vitro.
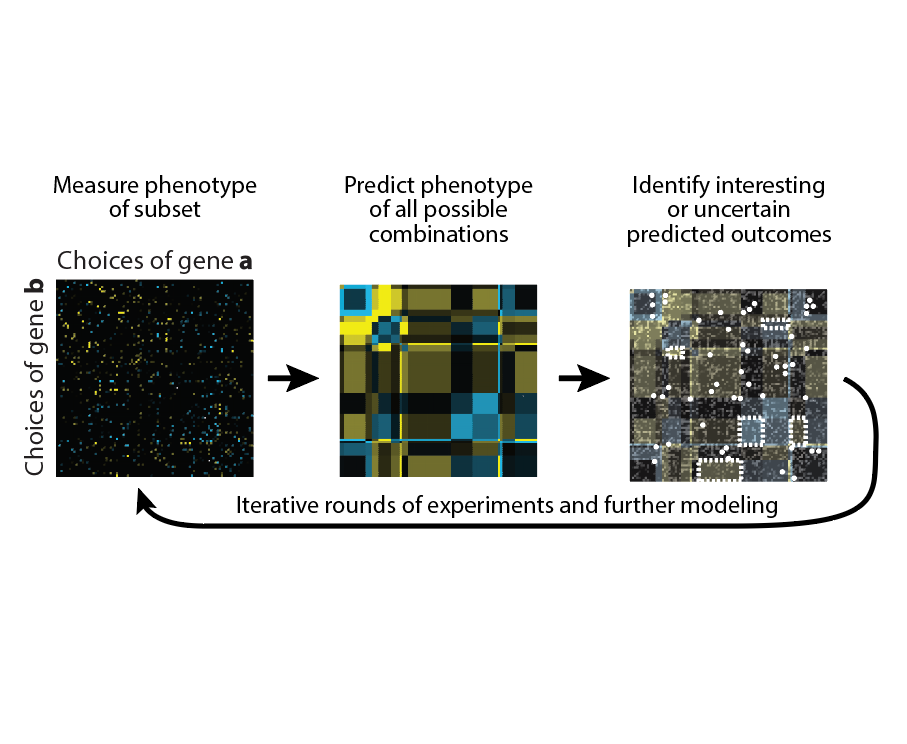
Combinatorial genetics
We are interested in developing new functional genomics approaches to tackle problems that are “beyond genome-scale.” For example, many interesting phenotypes likely arise through interactions between regulators, which are often far too numerous to measure exhaustively. Our lab is developing new experimental approaches for measuring genetic interactions and computational methods for prioritizing which ones to measure.
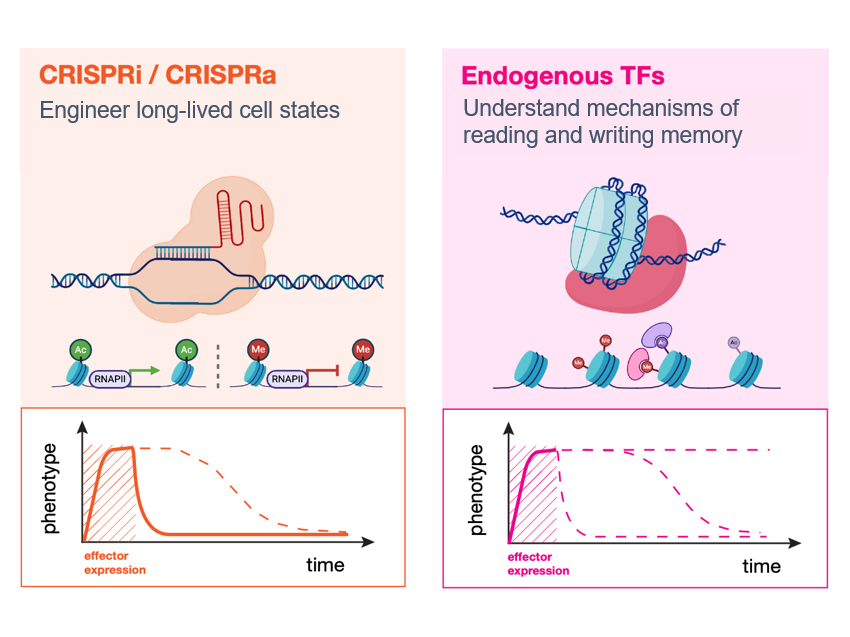
CRISPR tool development
We develop new CRISPR tools for perturbing the genome and measuring the consequences. A current interest is in methods for reading, writing, and quantifying epigenetic memory.